Optische Stöße
Dr. O. Hoffmann
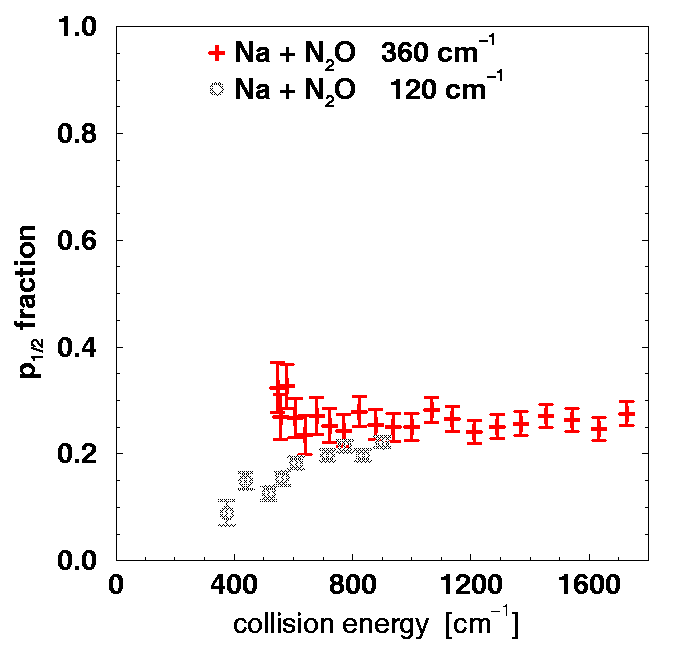
Endzustandsanalyse: Feinstrukturanteil
Natrium-Molekül-Stoßpaar
Die Situation bei Atom-Molekül-Stoßpaaren ist komplizierter
als bei Atom-Atom-Stoßpaaren.
Zum einen ist es schwieriger, die Potentialflächen präzise
zu berechnen. Zum anderen ist auch die Dynamikrechnung mit größeren
Schwierigkeiten behaftet. Umso interessanter ist es natürlich,
experimentelle Daten zur Verfügung zu stellen, um die verschiedenen
theoretischen Ansätze zu überprüfen.
Während für die theoretischen Daten zum Feinstrukturanteil bei den
Atom-Atom-Stoßpaaren quantenmechanische Dynamikrechnungen
durchgeführt wurden, wurden bei den Atom-Molekülstößen
klassische beziehungsweise semiklassische Rechnungen vorgenommen.
Insgesamt erfolgt die numerische Berechnung der theoretischen Vergleichsdaten
im wesentlichen in drei Schritten:
1. Quantenchemische Berechungen der Potentialflächen und der
Übergangsdipolmomente.
2. Klassische Trajektorienrechnungen (Die Molekül-Ausrichtung wird
mit einer Monte-Carlo-Methode variiert).
3. Die zeitabhängige Schrödingergleichung wird auf der
Trajektorie gelöst, um die nichtadiabatischen Übergangswahrscheinlichkeiten zu bestimmen.
Trotz der wesenlich komplizierteren Situation ergibt sich eine recht gute Übereinstimmung.